Adrenarquia prematura
Adrenarquia prematura
T. Gavela Pérez(1)(2)(3), F.J. Mejorado Molano (1), L.Guillén(1)(2)(3).
(1) Servicio de Pediatría, Hospital Universitario Fundación Jiménez Díaz. Madrid. (2) Unidad de Endocrinología Infantil, Hospital Universitario Fundación Jiménez Díaz. Madrid. (3) Laboratorio de Lípidos, Instituto de Investigación Sanitaria Fundación Jiménez Díaz. Madrid.
Fecha de recepción: 10 de septiembre 2017
Fecha de publicación: 30 de octubre 2017
Adolescere 2017; V (3): 31-42
Resumen
|
La aparición de forma temprana de vello púbico y/o axilar puede ser una simple variación de la normalidad o el reflejo de alguna patología que puede influir o condicionar el momento de su aparición. Aún sin patología de base es motivo de preocupación para los padres y pacientes y puede tener repercusiones negativas en la vida adulta. El pediatra de atención primaria es el primer profesional al que se consulta esta sintomatología y debe ser capaz de realizar un enfoque inicial adecuado, basado en una historia clínica y una exploración física exhaustiva, orientar a padres y pacientes y, en aquellos casos que considere necesario, derivar al paciente a un Servicio de Endocrinología Pediátrica. Palabras clave: adrenarquia prematura; pubarquia prematura; axilarquia prematura. |
Abstract
|
The age in which pubic and/or axillary hair appears varies greatly. Its early presentation may be a simple variation of normality or the manifestation of any of the multiple pathologies that may influence the moment of its appearance. In any case, even in the absence of disease, it is a reason for concern for the parents and patients and may have negative repercussions in the adult age. The primary healthcare pediatrician is the professional who initially faces this situation and who should perform an adequate diagnostic approach, guiding parents and patients, and in those cases where considered necessary, referring the patient to a Pediatric Endocrinology service. Key words: premature adrenarche; premature pubarche. |
Introducción
La glándula adrenal, encargada de la esteroidogénesis (Figura 1), está dividida en tres zonas: la zona fascicular que segrega glucocorticoides, la zona glomerular que produce mineralocorticoides y la zona reticular que produce andrógenos.
Durante la vida fetal esta glándula es un órgano grande y funcionalmente muy activo; por ello, las concentraciones de dehidroepiandrosterona sulfato (DHEA-S) en sangre de cordón son elevadas, pero tras el nacimiento la zona reticular de la glándula suprarrenal involuciona, proporcionando al organismo posteriormente niveles muy bajos de androstendiona y de DHEA(1). No obstante, esta glándula tiene un resurgir, fenómeno exclusivo de humanos y primates, caracterizado por un incremento de la producción de andrógenos, DHEA-S y 17-OH-progesterona, hecho conocido con el término adrenarquia.
La adrenarquia era considerada tradicionalmente como un acontecimiento brusco, que tenía lugar sobre los 6 años de edad para ambos sexos, pero en estudios posteriores utilizando métodos de alta sensibilidad, se ha visto que aparece de forma más progresiva ya desde los 3 años de vida. No se conoce con exactitud la fisiología de este fenómeno en el que se produce un aumento de la actividad de la enzima 17,20 liasa, que favorece el aumento de secreción de andrógenos. Parece que podrían influir factores metabólicos, genéticos y ambientales. Así, hay estudios que muestran edades más precoces de adrenarquia en niños con antecedente de bajo peso al nacimiento y, también, en niños que han presentado incrementos notables del índice de masa corporal en edades tempranas de la infancia (2,3), lo que deja relucir que el estado metabólico de los niños tiene una influencia franca sobre esta secreción hormonal. Además, se ha demostrado que la leptina, adipoquina secretada en el tejido adiposo y cuyos niveles sanguíneos se correlacionan de forma directa con la cantidad de masa grasa, aumenta la actividad de la enzima 17,20 liasa(4). Por otra parte, se ha constatado que el incremento de andrógenos circulantes objetivado durante la adrenarquia contribuye a favorecer los cambios de la composición corporal.
La gonadarquia, fenómeno independiente de la adrenarquia, es un fenómeno asociado a la maduración sexual y hace referencia al aumento brusco en la secreción de hormonas sexuales por parte de las gónadas (ovarios en las niñas y testículos en los niños)
La gonadarquia, fenómeno independiente de la adrenarquia, es un fenómeno asociado a la maduración sexual y hace referencia al aumento brusco en la secreción de hormonas sexuales por parte de las gónadas (ovarios en las niñas y testículos en los niños). La pubertad es un proceso biológico complejo en el que se alcanza el desarrollo de los caracteres sexuales secundarios, la capacidad reproductiva y la talla adulta. La pubertad está regulada por la interacción dinámica entre factores genéticos y ambientales desde edades tempranas de la vida. El incremento en el número y en la amplitud de los picos de secreción de hormona liberadora de gonadotropinas (GnRH) por parte de las neuronas hipotalámicas productoras de GnRH se ha propuesto como el fenómeno final que activaría la pubertad. Esta secreción hormonal, a su vez, incrementaría la producción de gonadotropinas y esteroides sexuales, favoreciendo el desarrollo de los caracteres sexuales secundarios (5,6). La adrenarquia generalmente precede en dos años a la gonadarquia y su mecanismo de control, como antes hemos citado, no se conoce exactamente.
La adrenarquia prematura se define como la aparición de vello púbico y/o vello axilar y/o incremento del olor corporal de origen apocrino antes de los 8 años en niñas y de los 9 años en niños y es fruto del exceso de precursores de andrógenos adrenales, principalmente DHEA-S
La adrenarquia prematura se define como la aparición de vello púbico y/o vello axilar y/o incremento del olor corporal de origen apocrino antes de los 8 años en niñas y de los 9 años en niños y es fruto del exceso de precursores de andrógenos adrenales, principalmente DHEA-S. La forma más frecuente de presentación de esta entidad es la adrenarquia prematura idiopática (API), cuyo diagnóstico es de exclusión tras descartar otras causas de exceso de producción de andrógenos tales como tumores adrenales o gonadales, hiperplasia adrenal congénita de presentación tardía o administración exógena de andrógenos (7,8). Por su forma de presentación clínica en ocasiones en la práctica habitual se usan de forma indistinta los términos adrenarquia prematura y pubarquia prematura, siendo esta última únicamente la manifestación clínica de presencia de vello púbico a una edad menor de 8 años en niñas y 9 años en niños (8).
Se desconoce exactamente la prevalencia de adrenarquia prematura en la población general (9), pero constituye un motivo de consulta relativamente frecuente en las consultas de pediatría, con una ratio mujer-varón alrededor de 9:1(10). Un estudio epidemológico, realizado en población estadounidense muestra en niñas pubarquia prematura hasta en un 15% de ellas (11).
Aunque la API se considera como una variante de la normalidad, esta entidad clínica ha sido relacionada con (8,12-16): a) antecedente pequeño para la edad gestacional (PEG); b) exceso de peso, alteraciones metabólicas e incremento de riesgo cardiovascular; c) síndrome de ovario poliquístico (SOP); d) adelanto puberal; d) repercusión de talla adulta. Con el objetivo de aclarar estas posibles asociaciones, nuestro grupo diseñó una cohorte de niñas con API en el año 2007 con el objetivo de realizar un seguimiento periódico de estas niñas desde el diagnóstico hasta alcanzar la edad de 18 años (17).
Para algunos autores, el SOP pudiera tener un origen prenatal pero que se manifiesta a partir de la adolescencia (8,13). En esta línea, una cohorte de niñas catalanas con el antecedente de pequeño para la edad gestacional (PEG) y API, progresa en un 45% a SOP en etapas posteriores (14).
Una explicación fisiológica que trata de justificar la relación entre PEG y API es que la desnutrición en la etapa prenatal condicionaría una serie de cambios epigenéticos que alterarían la funcionalidad de la glándula adrenal tras el nacimiento. A este hecho habría que sumar la potencial influencia de la velocidad de recuperación de peso y talla en la etapa posnatal (8,18). No obstante, no hay evidencias concluyentes sobre que el antecedente de PEG pueda predisponer directamente a desarrollar API (17). Esto podría deberse a que los estudios se han diseñado en poblaciones diferentes, con distintos tamaños muestrales y con distintos criterios de definición de API y PEG (8,16). Asimismo, tampoco se ha encontrado asociación entre exceso de peso y alteraciones metabólicas con API en algunas investigaciones (17).
Otra línea de investigación con resultados contradictorios (16) es la asociación de API con adelanto puberal y repercusión sobre talla final. En esta línea, nuestro grupo de trabajo (datos pendientes de publicar) ha objetivado cierto adelanto puberal en las niñas con API sin evidenciar repercusión de la talla final con respecto a su talla genética.
En definitiva, los datos procedentes de distintas investigaciones sobre las consecuencias a largo plazo de la API no son unánimes. Por ello, se plantea la necesidad de diseñar estudios longitudinales multicéntricos, que engloben distintas áreas geográficas, con criterios homogéneos de diagnóstico de API, para evaluar su relación con PEG, SOP, incremento de riesgo cardiovascular, adelanto puberal y repercusión sobre la talla final.
Manifestaciones clínicas
Para orientar el diagnóstico etiológico es importante conocer la rapidez de la aparición de las manifestaciones clínicas junto a la evolución de la velocidad de crecimiento y la talla
Aparición de vello púbico y/o vello axilar y/o aumento del olor corporal de origen aprocino antes de los 8 años en las niñas y de los 9 años en los niños, sin que esta sintomatología se acompañe de desarrollo mamario en las niñas ni aumento del volumen testicular en los niños.
Junto a los datos clínicos anteriormente comentados, será de suma utilidad para orientar el diagnóstico etiológico conocer la rapidez de la aparición de las manifestaciones clínicas junto a la evolución de la velocidad de crecimiento y la talla.
Etiopatogenia
1. Adrenarquia prematura idiopática: forma más frecuente. Constituye un diagnóstico de exclusión. Esta forma clínica evoluciona lentamente, asociando una velocidad de crecimiento moderadamente acelerada, sin asociar datos de desarrollo puberal. En esta entidad clínica las niñas y niños suelen presentar una talla alta para su edad cronológica pero concordante con su edad ósea que suele estar aumentada no más de 2 SDS respecto a edad y sexo, con predicciones de talla acordes a su talla diana en el momento del diagnóstico. En este grupo, podemos encontrarnos con una forma de presentación denominada adrenarquia exagerada en la que se evidencia mayor aceleración del crecimiento y adelanto de la edad ósea. La prevalencia de insulinorresistencia y de evolución a síndrome de SOP es mayor en este último grupo.
2. Formas tardías, no clásicas, de hiperplasia suprarrenal congénita: enfermedades de origen genético y de herencia autosómica recesiva caracterizadas por déficit enzimáticos en la biosíntesis del cortisol. Este déficit de cortisol produce, por un mecanismo de retroalimentación negativa, un aumento de la producción de hormona adrenocorticotropa (ACTH) y secundariamente una hiperestimulación de la corteza suprarrenal motivando una elevación de los esteroides previos al bloqueo enzimático. Las formas clásicas se diagnostican de forma temprana, tanto por su clínica (genitales ambiguos en las niñas, signos francos de virilización precoz en los niños, y en algunos casos asocian HTA, hipopotasemia…), como por su detección en el cribado metabólico realizado de forma sistemática a las 48 horas de vida. Dentro de ellas destacamos:
Las formas clásicas se diagnostican de forma temprana, tanto por su clínica (genitales ambiguos en las niñas, signos francos de virilización precoz en los niños, y en algunos casos asocian HTA, hipopotasemia…), como por su detección en el cribado metabólico realizado de forma sistemática a las 48 horas de vida
a. Deficiencia de 21-α-hidroxilasa (CYP21A2): la forma más común y que se sospecha objetivando valores aumentados de 17-OH- progesterona basal y tras estímulo con ACTH. El diagnóstico de confirmación se realiza mediante el oportuno estudio genético.
b. Deficiencia de 11-β-hidroxilasa (CYP11B1): forma excepcional. A diferencia de la forma clásica en este caso no se asocia a HTA e hipopotasemia. Los valores basales y tras estímulo con ACTH de 11-desoxicortisol se encuentran muy elevados.
c. Deficiencia de 3-β-hidroxiesteroide deshidrogenasa (HSD3B2): forma excepcional. Se presenta con niveles aumentados de DHEA-S, pero no en rango tumoral, lo que en ocasiones es difícil distinguir de la API. Los niveles sanguíneos de 17-OH-pregnenolona, basales y tras estímulo, aparecen muy incrementados.
3. Hiperandrogenismos adrenales congénitos ACTH dependientes:
a. Resistencia a glucocorticoides: defecto en la señalización del receptor de glucocorticoide, existiendo una retroalimentación negativa deficiente y consecuentemente una respuesta compensatoria de ACTH.
b. Deficiencia de cortisona reductasa: defecto en el metabolismo periférico del cortisol. Es un fallo en la regeneración de cortisol desde cortisona lo que conduce a un aumento compensatorio de ACTH y un hiperandrogenismo adrenal moderado. Está causado por el déficit del enzima 11-β-hidroxiesteroide deshidrogenasa tipo 1 (HSD11B1), que es funcionalmente la mejor cortisona reductasa, o por la deficiencia de hesoxa-6-fosfato-deshidrogenasa (H6PDH) que es un cofactor de la cortisona reductasa.
4. Otros hiperandrogenismos adrenales:
a. Defecto en la sulfatación de DHEA. No afecta a la síntesis de corticoesteroides. Se trata de una mutación en el gen PAPSS2 (3-fosfoadenosis-5-fosfosulfatosintasa) que codifica el donante de sulfato al SULT2A1, por ello parece una deficiencia de este último gen. Se caracteriza por bajos niveles sanguíneos de DHEA-S, pero aumenta la conversión de DHEA a andrógenos activos como androstendiona.
5. Tumores virilizantes: de origen gonadal (ovárico en niñas y testicular en niños), suprarrenal o tumores germinales productores de β-HCG extragonadales (hígado, pulmón, cerebro). Son signos de sospecha de estas patologías un rápido incremento del vello púbico/axilar/corporal acné quístico, velocidad de crecimiento y edad ósea sumamente aceleradas, incremento de la musculatura, voz grave, clitoromegalia o crecimiento exagerado del pene en el niño. Dependiendo de la naturaleza del tumor de origen suprarrenal podemos encontrarnos signos de hipercortisolismo.
Diagnóstico diferencial
1. Vello fino no terminal en área púbica y/o axilar.
2. Hipertricosis: incremento del vello corporal de forma anormal para edad, raza y sexo en zonas no típicamente androgénicas.
La Hipertricosis consiste en el incremento del vello corporal de forma anormal para edad, raza y sexo en zonas no típicamente androgénicas
3. Iatrógena: fármacos que incrementen el vello corporal.
4. Pubertad precoz: auténtico reto diagnóstico, particularmente en niños. Como se ha comentado previamente, la API es mucho más frecuente entre las niñas. De esta forma, la presencia de adrenarquia prematura en un niño con tamaño testicular inferior a 4 ml nos obliga a descartar una pubertad precoz periférica (tumores virilizantes de origen adrenal, testicular o, más excepcionalmente, productores de β-HCG). La presencia de tamaño testicular igual o superior a 4 ml orienta a una pubertad precoz de origen central. En el caso de las niñas, la presencia de adrenarquia prematura junto a telarquia (antes de los 8 años) nos obliga a descartar la presencia de pubertad precoz. Es preciso reseñar que, en el transcurso normal de la pubertad en las niñas, hasta en 25% de los casos la pubarquia precede a la aparición de telarquia.
La presencia de adrenarquia prematura en un niño con tamaño testicular inferior a 4 ml nos obliga a descartar una pubertad precoz periférica (tumores virilizantes de origen adrenal, testicular o, más excepcionalmente, productores de β-HCG)
Evaluación diagnóstica
La evaluación diagnóstica de estos pacientes conlleva una anamnesis completa, con énfasis en determinados aspectos, una exploración física rigurosa y una serie pruebas complementarias orientadas al diagnóstico etiológico (Tabla 1).
Aunque la API es la forma más frecuente de adrenarquia prematura y se considera una variante de la normalidad, constituye un diagnóstico de exclusión. Por ello, es absolutamente recomendable derivar a niños/as con sospecha de adrenarquia prematura desde atención primaria a atención especializada. Con mayor celeridad en el caso de los niños, donde las posibilidades de presentar patología más grave se incrementan notablemente.
La presencia de tamaño testicular igual o superior a 4 ml orienta a una pubertad precoz de origen central
Anamnesis
Historia actual: describir con precisión el tiempo de evolución y la rapidez de instauración de pubarquia y/o axilarquia y/o aumento del olor corporal de origen apocrino junto con otras manifestaciones tales como la presencia de acné, pelo graso y aumento del vello corporal. Otros datos primordiales de los que será preciso obtener información son la evolución de la velocidad de crecimiento, talla e índice masa corporal. En el caso de las niñas, de forma específica, demandaremos información relativa a la presencia de otros datos de hiperandrogenismo como la presencia de clitoromegalia. Asimismo, deberemos preguntar por la aparición de telarquia, así como por la presentación de sangrado vaginal. En los niños, interrogaremos de forma específica sobre el incremento del tamaño del pene, de la bolsa escrotal y cambio en la voz.
En las chicas, la presencia de adrenarquia prematura junto a telarquia (antes de los 8 años) nos obliga a descartar la presencia de pubertad precoz
Antecedentes familiares: hay que recoger información relativa a raza, país de origen, datos del desarrollo puberal de los padres, antecedentes familiares de hirsutismo, irregularidades menstruales, problemas de fertilidad y SOP.
Antecedentes personales: raza, país de origen, embarazo, datos perinatales (edad gestacional y datos antropométricos), enfermedad y/o terapia crónica.
Exploración física
Datos antropométricos: peso, talla, IMC y velocidad de crecimiento en valor absoluto, percentiles y SDS para tablas de referencia.
La API es la forma más frecuente de adrenarquia prematura y se considera una variante de la normalidad, constituye un diagnóstico de exclusión
Fenotipo: aspecto “cushingoide”.
Tensión arterial: en valor absoluto y en percentiles para tablas de referencia.
Piel: presencia de acné, estrías rojo-vinosas, acantosis nigricans, exceso de vello corporal (hirsutismo en las niñas).
Abdomen: descartar viseceromegalias y distensión abdominal.
Vello: comprobar realmente si se existe vello terminal en área púbica y axilar.
Niñas: describir con detalle la presencia de vello y forma del mismo en pubis y axilas. Valorar tamaño de clítoris. Descartar la presencia de telarquia.
Niños: describir con detalle la presencia de vello y forma del mismo en pubis y axilas. A continuación, valorar si existe asimetría testicular. Seguidamente, deberemos valorar el tamaño testicular (menor, igual o mayor a 4 ml de Prader). Finalmente, cuantificaremos el tamaño del pene y tendremos en cuenta tablas de referencia para la edad.
Pruebas complementarias
a. Pruebas de imagen:
Edad ósea: debería solicitarse de inicio en todos los casos. Si está muy adelantada (más de 2 SDS para edad y sexo) sugiere un exceso de secreción de andrógenos.
Se realizara ecografia testicular ante la presencia de asimetría testicular
Ecografía abdominal: ante la sospecha de tumor de origen suprarrenal. Clínica rápidamente progresiva junto con valores muy elevados de DHEA-S. De forma excepcional, ante sospecha de tumor germinal productor de β-HCG.
Ecografía pélvica: para descartar tumor ovárico (en caso de duda la prueba de imagen de elección sería la RM). Obligado realizar ante clínica rápidamente progresiva y/o aumento notable de los niveles circulantes de andrógenos. También nos puede mostrar datos de impregnación estrogénica (tamaño uterino y ovárico, relación cuello/fundus uterino, presencia de línea endometrial) en caso de niñas que tengamos dudas sobre si ha comenzado la pubertad (ejemplo: niña obesa en la que es difícil discernir entre adipomastia y verdadera telarquia).
Ecografía testicular: de entrada, ante la presencia de asimetría testicular. También, testículos simétricos < 4 ml con marcada elevación de testosterona.
La testosterona es de síntesis prácticamente exclusiva en testículos y ovarios. Valores muy elevados > 2 ng/ml) sugieren patología tumoral
b. Analítica de sangre basal que debe incluir:
Andrógenos basales:
— DHEA-S: de síntesis exclusivamente suprarrenal. En la API encontraremos valores ligeramente elevados para estadio puberal Tanner I. Los valores de DHEA-S superiores a 700 µg/dl son sugerentes de patología tumoral.
— Androstendiona: se sintetiza en glándula suprarrenal pero también en ovario y testículo. En la API los valores son normales. En formas de hiperplasia suprarrenal tardía encontraremos niveles sanguíneos ligeramente elevados.
— Testosterona: de síntesis prácticamente exclusiva en testículos y ovarios. Valores muy elevados (> 2 ng/ml) sugieren patología tumoral.
— 17-OH-progesterona: síntesis mixta, principalmente en glándula suprarrenal y en menor medida en el ovario. En teoría, valores por debajo de 2 ng/ml descartarían formas de hiperplasia suprarrenal de inicio tardío.
Se realizara el estudio del cortisol libre urinario de 24 h, cuando haya sospecha clínica de hipercortisolismo (adrenarquia prematura + exceso de peso + hipertensión arterial + fenotipo característico)
Metabolismo hidrocarbonado y perfil lipídico, sobre todo en los casos que asocian exceso de peso y antecedentes de bajo peso para edad gestacional.
Función tiroidea
c. Prueba de orina:
Estudio de cortisol libre urinario de 24 horas: cuando sospecha clínica de hipercortisolismo (adrenarquia prematura + exceso de peso + hipertensión arterial + fenotipo característico).
d. Pruebas funcionales hormonales:
Test de estimulación de hormona adrenocorticotropa (ACTH) para 17-OH-progesterona y cortisol: es de utilidad para el diagnóstico etiológico de hiperplasia suprarrenal congénita de presentación tardía. Debe realizarse a primera hora de la mañana (entre las 08:00 y las 09:00 horas), extrayendo en el momento 0 cortisol y 17-OH-progesterona para, posteriormente, administrar 250 microgramos (µg) de ACTH. Seguidamente se extraerán muestras de sangre para determinar cortisol y 17-OH-progesterona a los 30 y 60 minutos. Para considerar la prueba válida el pico de cortisol ha de ser superior a 20 µg /dl. Existe controversia en cuanto a la cifra de 17-OH-progesterona a partir de la cual considerar una alta sospecha de hiperplasia suprarrenal congénita, aunque el criterio internacional más extendido es un pico superior a 15 ng/ml. No obstante, el diagnóstico de confirmación es genético y pruebas en las que los picos de 17-OH-progesterona se sitúen entre 10 y 15 ng/ml también sería recomendable la realización de un estudio genético.
Otro motivo de controversia es la realización de entrada de un test de ACTH a todas las niñas con sospecha de adrenarquia prematura o sólo a las que presenten edad ósea adelantada y elevación basal de 17-OH-Progesterona. En nuestra opinión debería generalizarse la realización del test de ACTH ante una niña con adrenarquia prematura por los siguientes motivos: a) existen formas paucisintomáticas de hiperplasia suprarrenal congénita (sin adelanto de la edad ósea, velocidad de crecimiento normal, 17-OH-progesterona normal o moderadamente elevada); b) enorme variabilidad de los valores de 17-OH-progesterona en función de la hora del día a la que se determinan sus concentraciones; c) si el estudio basal muestra unos valores elevados de 17-OH-progesterona, es necesario realizar con posterioridad un test de ACTH. No obstante, será preciso individualizar cada paciente y otra opción razonable sería la realización de una analítica basal inicial, teniendo en cuenta las salvedades anteriormente comentadas, con un estrecho seguimiento del paciente.
Test de estimulación de hormona adrenocorticotropa (ACTH) para 17-OH-pregnenolona y 11-desoxicortisol: considerar en los casos de adrenarquia exagerada (edad ósea muy adelantada junto con marcada aceleración de la velocidad de crecimiento) en el que el estudio anterior es normal para descartar formas tardías de déficit de 11-β-hidroxilasa y 3-β-hidroxiesteroide deshidrogenasa
Test de supresión con dexametasona: ante cuadros virilizantes progresivos puede ser útil esta prueba para diferenciar principalmente tumores virilizantes, en los que no habría supresión, de otras formas de virilización como HSC o adrenarquia exagerada en los que sí habría respuesta supresora de andrógenos con dexametasona. No obstante, pensamos que en la mayoría de ocasiones podría ser suficiente con la cuantificación de andrógenos basales (muy elevados en los casos de tumores) y la realización de pruebas de imagen.
e. Estudios genéticos:
Orientados al diagnóstico etiológico: fundamentalmente cuando exista sospecha de hiperplasia congénita de presentación tardía (estudio de gen CYP21A2).
Fase de investigación: el término idiopático indica causa desconocida en el momento actual. Pensamos que paralelamente a los avances genéticos de los próximos años, especialmente con el desarrollo de las nuevas técnicas de secuenciación masiva, iremos conociendo las bases moleculares de la API.
Se proponen los algoritmos diagnósticos mostrados en la figura 2 para las niñas y en la figura 3 para los niños. No obstante, debe individualizarse cada paciente tanto para el diagnóstico como para el seguimiento.
Tratamiento
En el caso que sea posible debe ser etiológico: los tumores deben ser extirpados. Ante una hiperplasia suprarrenal congénita de presentación tardía en un niño prepuberal se tendrá en cuenta su expresividad clínica (edad ósea, talla, pronóstico de talla final, acné…) para valorar la administración de corticoterapia oral en forma de hidroaltesona hasta la finalización del crecimiento con la dosis mínima que sea capaz de controlar la sintomatología y evitar efectos secundarios (hipercortisolismo iatrógeno). Es un tema complejo que se escapa a los objetivos principales de esta revisión.
La API no requiere un tratamiento médico específico pero a nuestro juicio sí es recomendable:
— Seguimiento periódico de forma semestral al menos los dos primeros años del diagnóstico con un doble objetivo: a) vigilar nuevos signos de virilización y aceleración del crecimiento causados por una patología no detectada en análisis preliminares; b) descartar adelanto puberal.
— La diferencia entre el aspecto físico con los otros niños de su edad y con el desarrollo cognitivo y recursos emocionales propios de la edad pueden ser negativos desde el punto de vista psicológico en estos pacientes, por lo que hay que considerar esta vulnerabilidad y valorar apoyo psicológico, la mayoría de las veces desde su entorno familiar y, en ocasiones, con psicólogos infantiles.
— Para el tratamiento de los síntomas que pueden incomodar tanto a los niños como a su entorno, se pueden usar desodorantes exentos de metales como la piedra de alumbre y depilar el vello con métodos poco agresivos para la piel.
— Modificación del estilo de vida y ejercicio físico regular. En situaciones de exceso de peso se tomarán medidas destinadas a alcanzar un IMC dentro de la normalidad. Hay que recordar que en algunas series se ha relacionado la API con incremento de riesgo cardiovascular.
— Tras el comienzo de la pubertad, se recomienda seguimiento periódico de al menos una vez al año de adolescentes con API por elevada prevalencia posterior de desarrollo de SOP, sobre todo en aquellas con antecedente de adrenarquia exagerada y exceso de peso. Por ello, será muy importante trabajar en medidas preventivas tales como asesoramiento dietético, modificación de estilo de vida y fomentar el ejercicio físico. Si a pesar de estas medidas persiste clínica muy florida, principalmente hirsutismo, con importante repercusión psicológica, aparte de la implementación de medidas estéticas será necesario valorar terapia médica: a) anticonceptivos orales con acción antiandrogénica; b) metformina: pacientes en las que a su vez se objetiva exceso de peso e insulinorresistencia con otras alteraciones metabólicas. Indicación fuera de la guía práctica clínica habitual en la que hay que obtener el consentimiento de la paciente y familiar pero con resultados alentadores en este grupo concreto de pacientes (19).
Tras el comienzo de la pubertad, se recomienda seguimiento periódico de al menos una vez al año de adolescentes con API por elevada prevalencia posterior de desarrollo de SOP, sobre todo en aquellas con antecedente de adrenarquia exagerada y exceso de peso
Conclusión
En definitiva, la adrenarquia prematura se define como la presencia de vello púbico y/o axilar y/o incremento del olor corporal de origen apocrino antes de los 8 años en las niñas y de los 9 años en los niños. La causa más frecuente es la API; sin embargo, requiere previamente de un diagnóstico de exclusión de otros cuadros causantes de hiperandrogenismo como por ejemplo la hiperplasia suprarrenal de presentación tardía o tumores virilizantes localizados en glándula adrenal, ovario y testículo. Aunque la API se considera una variante de la normalidad, existe controversia sobre su asociación con antecedente de PEG, SOP, exceso de peso, riego cardiovascular, adelanto puberal y repercusión sobre talla adulta. Por ello, se plantea un seguimiento periódico de estos niños/as hasta alcanzar la edad adulta.
En la actualidad existe controversia sobre la asociación de la API con antecedente de PEG, SOP, exceso de peso, riego cardiovascular, adelanto puberal y repercusión sobre la talla adulta
Tablas y figuras
Tabla 1. Anamnesis, examen físico, pruebas complementarias y diagnóstico diferencial de la adrenarquia prematura
|
ANAMNESIS |
EXAMEN FÍSICO |
PRUEBAS COMPLEMENTARIAS |
|
Historia actual Tiempo de evolución Rapidez de instauración ¿Presencia acné, pelo graso y vello corporal? Gráficas de crecimiento Niñas: — ¿Telarquia? — ¿Sangrado vaginal? Niños: — ¿Tamaño pene? — ¿Cambios en la voz? |
Datos antropométricos • Peso (kg y SDS) • Talla (kg y SDS) • IMC (kg/m2 y SDS) • Velocidad de crecimiento (cm/año y SDS) Fenotipo (aspecto Piel: estrías, hirsutismo, Palpación abdominal Comprobar tipo de vello o Niñas: descartar telarquia y clitoromegalia o Niños: descartar |
Pruebas de imagen • Edad ósea: de inicio en todos los casos • Ecografía testicular: de inicio si • Ecografía abdominal: descartar • Ecografía pélvica: descartar tumor ovárico (clínica progresiva + ↑↑andrógenos) |
|
Antecedentes personales Raza, país de origen Embarazo Datos perinatales Enfermedades crónicas Tratamientos recibidos |
Analítica sanguínea Andrógenos basales: • DHEA-S • Androstendiona • Testosterona • 17-OH-progesterona Metabolismo hidrocarbonado y perfil lipídico Función tiroidea |
|
|
Antecedentes familiares Datos de desarrollo Hirsutismo y/o SOP. |
Cortisol libre urinario de 24h: Pruebas funcionales hormonales • Test de ACTH para 17-OH-progesterona y cortisol: descartar formas tardías HSC por déficit de 21-α-hidroxilasa • Test de ACTH para 17-OH-pregnenolona y 11-desoxicortisol: en adrenarquia exagerada, cuando test para 17-OH-progesterona normal • Test de supresión con dexametasona: individualizar en cuadros virilizantes importantes Estudios genéticos orientados |
|
|
DIAGNÓSTICO DIFERENCIAL |
||
|
• Vello fino no terminal en área púbica y/o axilar • Hipertricosis • Adrenarquia iatrógena • Pubertad precoz |
||
Figura 1. Esquema de la esteroideogénesis
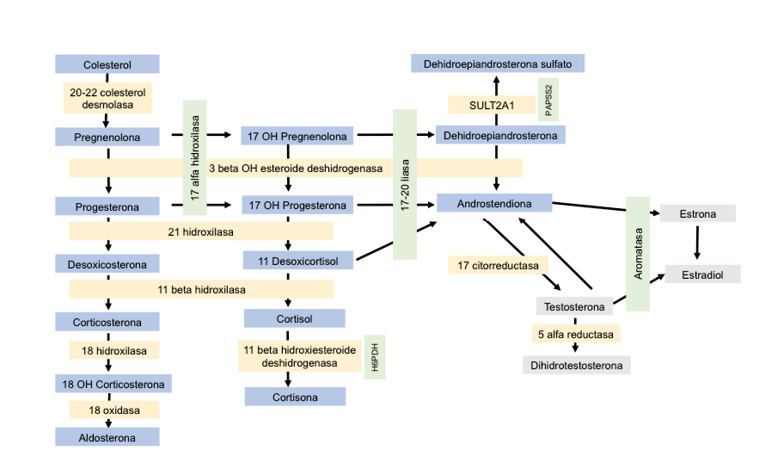
H6PDH: hexosa-6-fosfato deshidrogenasa; gen PAPSS2
Figura 2. Algoritmo de adrenarquia prematura en niñas
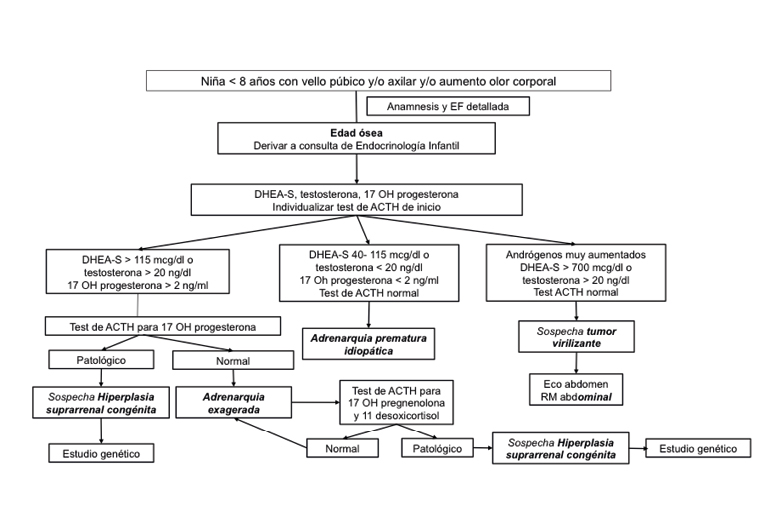
Figura 3. Algoritmo de adrenarquia prematura en niños
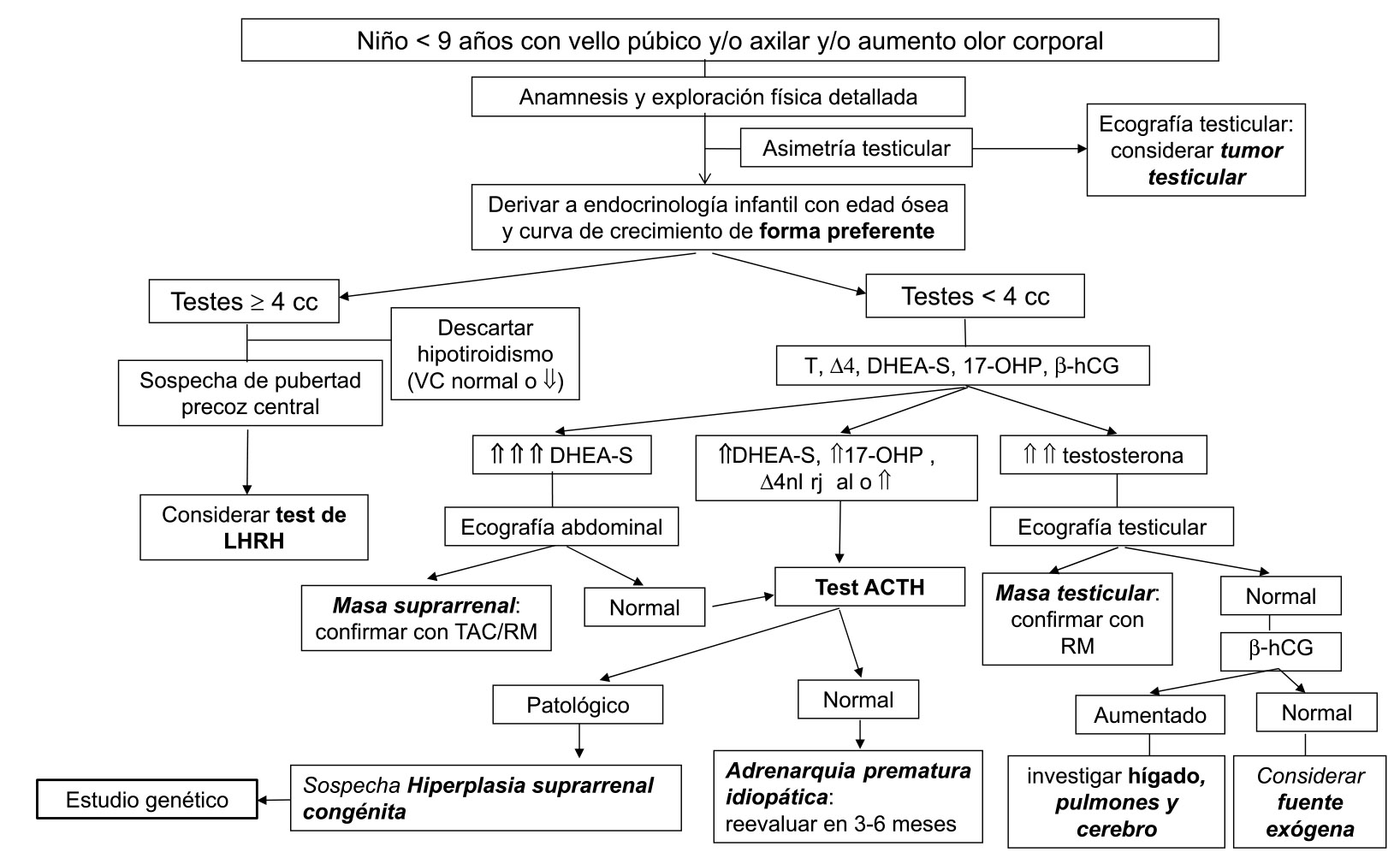
RM: resonancia magnética
Bibliografía
1. De Peretti E, Forest MG. Unconjugated dehydroepiandrosterone plasma levels in normal subjects from birth to adolescence in human: the use of a sensitive radioimmunoassay. J Clin Endocrinol Metab. 1976; 43:982-91.
2. Marakaki C, Karapanou O, Gryparis A, Hochberg Z, Chrousos G, Papadimitriou A. Early Adiposity Rebound and Premature Adrenarche. J Pediatr 2017; 186:72-7
3. Ibáñez L, Potau N, Marcos MV, de Zegher F. Exaggerated adrenarche and hyperinsulinism in adolescent girls born small for gestational age. J Clin Endocrinol Metab. 1999; 84: 4739-41.
4. Biason-Lauber A, Zachmann M, Schoenle J. Effect of leptin on CYP 17 enzymatic activities in human adrenal cells: new insight in the onset of adrenarche. Endocrinology 2000; 141:1446-54
5. Ojeda S, Dubay C, Lomniczi A, Kaidar G, et al. Gene networks and the neuroendocrine regulation of puberty. Mol Cell Endocrinol 2010; 324: 3-11
6. Pozo J, Marquez M, Muñoz MT. Pubertad precoz y retraso puberal. Adolescere 2017; V: 23-49
7. Voutilainen R, Jääskeläinen J. Premature adrenarche: Etiology, clinical findings, and consequences. J Steroid Biochem Mol Biol. 2015; 145:226-36.
8. Idkowiak J, Lavery GG, Dhir V, Barrett TG, Stewart PM, Krone N, et al. Premature adrenarche: Novel lessons from early onset androgen excess. Eur J Endocrinol. 2011; 165:189-207.
9. Silverman SH, Migeon C, Rosemberg E, Wilkins L. Precocious growth of sexual hair without other secondary sexual development; premature pubarche, a constitutional variation of adolescence. Pediatrics. 1952; 10:426-32.
10. García Cuartero B. Pubarquia, adrenarquia, hirsutismo. Pediatr Aten Primaria. 2009; 11:143-54.
11. Herman-Giddens ME, Slora EJ, Wasserman RC, Bourdony CJ, Bhapkar MV, Koch GG, Hasemeier CM. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics. 1997; 99:505-12.
12. Rosenfield RL. Clinica review: Identifying children at risk for polycystic ovary síndrome. J Clin Endocrinol Metab 2007; 92:787-96
13. Connor EL. Adolescent polycystic ovary syndrome. Adolesc Med State Art Rev. 2012; 23:164-77.
14. Ibáñez L, Díaz R, López-Bermejo A, Marcos MV. Clinical spec- trum of premature pubarche: Links to metabolic syndrome and ovarian hyperandrogenism. Rev Endocr Metab Disord. 2009; 10:63-76.
15. Dhatariya K, Bigelow ML, Nair KS. Effect of dehydroepiandroste- rone replacement on insulin sensitivity and lipids in hypoadrenal women. Diabetes. 2005; 54:765-9.
16. Williams RM, Ward CE, Hughes I. Premature adrenarche. Arch Dis Child. 2011; 97:250-4.
17. Mejorado Molano FJ, Andrés Zallo L, Fornos Rodríguez M, Pérez Segura P, Gavela Pérez T, Sanz Calvo ML, Soriano Guillén L. The relationship between metabolic disorders and small for gestational age with idiopathic premature adrenarche. An Pediatr (Barc). 2016 [Epub ahead of print]
18. Mericq V. Low birth weight and endocrine dysfunction in post- natal life. Pediatr Endocrinol Rev. 2006; 4:3-14.
19. Williams T, Mortada R, Porter S. Diagnosis and Treatment of Polycystic Ovary Syndrome. Am Fam Physician 2016; 84:106-13

