Síndrome Poliuria-Polidipsia e Hipocrecimento
Síndrome Poliuria-Polidipsia e Hipocrecimento
M.T. Muñoz Calvo.
Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Departamento de Pediatría. Universidad Autónoma. Madrid.
Adolescere 2017; V (2): 99-103
Resumen
|
Se presenta un caso de una niña de 8 años y 10 meses con un cuadro de poliuria-polidipsia de un mes de evolución, junto con hipocrecimiento. Se realizan controles periódicos de imagen así como estudios hormonales del eje hipotálamo-hipofisario siendo normales. Al año de seguimiento presenta un incremento del tallo hipofisiario que confirma el diagnóstico de germinoma puro. Se realiza tratamiento con radioterapia y tras dos años de seguimiento con resonancias normales, se instaura tratamiento con hormona de crecimiento.. Palabras clave: Poliuria; Polidipsia; Tallo hipofisario; Resonancia craneal; Radioterapia. |
Abstract
|
We present a case of a girl of 8 years and 10 months with a polyuria and polydipsia of a month of duration associated a growth failure. MRI brain and hypothalamus-hypophysis hormone studies were normal. She was diagnosed of idiopathic central diabetes insipidus. After a year of follow-up, MRI brain showed thickened pituitary stalk. Diagnosis of germinoma was made. Radiotherapy were performed with adequate evolution. After two years of follow-up growth hormone deficit was diagnosed and treatment. Key words: Poliuria; Polidipsia; Tallo hipofisiario; Resonancia craneal; Radioterapia |
Motivo de consulta
Niña de 8 años y 10 meses de edad que acude al Servicio de Urgencias por cuadro de poliuria, polidipsia y nicturia de inicio brusco hace 30 días. No ha presentado cambios en el comportamiento, pero la sintomatología le impide la actividad cotidiana y desempeño escolar. Asimismo, notan sus padres que desde hace un año presenta una disminución del ritmo de crecimiento. Sin cefalea, vómitos ni trastornos de visión.
Antecedentes Familiares: madre: talla 155 cm, menarquia 13 años, padre: talla 177 cm, desarrollo puberal normal. Talla genética: 159 ± 5cm. Antecedentes Personales: embarazo y parto normal. PRN: 3000 g. LRN: 50 cm. Sin antecedentes de interés hasta la enfermedad actual.
Exploración física: Peso: 24 Kg (P 10-25), Talla: 126 cm (P25), TA: 89/65, FC: 90 l/m, Tª 36,7ºC Sin signos de deshidratación, ni rasgos dismórficos. Tanner I (T1, P1, Aa). Avidez por el agua, sin especial apetencia por líquidos fríos.
Estudios complementarios
► Hemograma: normal
► Bioquímica: – Glucemia: 94 mg/dL – Na+: 139 mEq/L – K+: 3,4 mEq/L – Cl–: 111 mEq/L
► Osmolaridad suero: 295 mOsm/kg H2O ► Osmolaridad orina: 70 mOsm/kg H2O
Ante la sospecha de diabetes insípida central, se realiza test de restricción hídrica que confirma el diagnóstico:
Tras 10 horas de restricción hídrica:
• Na+: 149 mEq/L. Osmols: 350 mOsm/kg/H20. Osmolo: 200 mOsm/kg/H20.
• Concentrando en orina hasta 537 mOsm/kg tras la administración de vasopresina.
• Se inicia tratamiento con desmopresina oral (60 mcg/día), con buen control posterior.
Se realiza a continuación: Estudio hipofisario: normal y RM: Ausencia neurohipófisis/tallo normal. Se realizan controles periódicos cada 6 meses.
A los 10 años y 11 meses, se realiza nueva RM craneal, objetivándose un engrosamiento del tallo hipofisario (4-5 mm) y glándula hipofisaria normal. Asimismo, presenta niveles muy disminuidos de IGF-I e IGFBP-3, sugerentes, en el contexto clínico de deficiencia de hormona de crecimiento (GH), experimentando una disminución importante de la velocidad de crecimiento, con retraso de la edad ósea (EO) de aproximadamente 2 años. Se realiza test de clonidina e hipoglucemia insulínica, que confirma el diagnóstico de deficiencia de GH.
Controles oftalmológicos, ecografía abdominal, serie ósea (sin lesión osteolítica) y determinación de marcadores tumorales en LCR (alfa-fetoproteína y βHCG) normales.
Los controles periódicos de RM craneal muestran un aumento progresivo del engrosamiento del tallo hipofisiario, de hasta 17x22x20 mm, realizándose biopsia con el diagnóstico de germinoma puro. Se inicia tratamiento con radioterapia fraccionada (40 Gy durante 4-5 semanas). Posteriormente se realiza RNM craneal seriada, sin observarse signos de recidiva tumoral ni diseminación leptomeníngea. Tallo hipofisiario fino y centrado.
Evolución clínica
1. Déficit de ADH.- Desde su diagnóstico a la edad de 8 años y 10 meses, se encuentra en tratamiento con desmopresina oral. En la actualidad, recibe una dosis de 60 mcg c/12 horas.
4. Déficiencia de GH.- Desde que fue diagnosticado de su tumoración supraselar, los niveles séricos de IGF-I e IGFBP-3 se han mantenido muy disminuidos, lo que en el contexto de su deficiencia hipofisaria múltiple sugiere una deficiencia de GH.
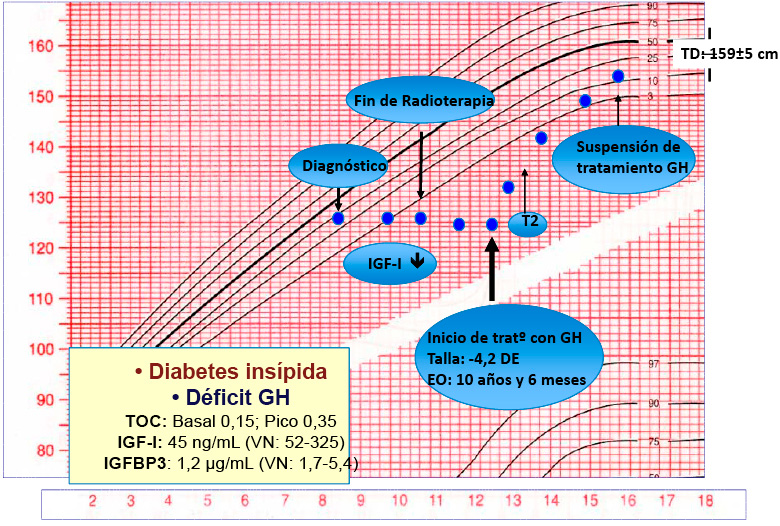
A los 12 años y 9 meses de edad inicia tratamiento con hormona de crecimiento recombinante, presentando una respuesta adecuada sin efectos secundarios de interés (Figura). A los 13 años y 4 meses y con una edad ósea de 11 años y 6 meses inicia el desarrollo puberal, presentando una evolución normal y menarquia a los 15 años y 7 meses (Figura 1). Actualmente la paciente tiene una edad cronológica de 17 años y 9 meses. Presenta una talla de 159,3 cm (P 25-50), normal para su talla genética y asimismo, presenta ciclos menstruales regulares.
Diagnóstico. 1.Germinoma puro (tratado con radioterapia) 2. Deficiencia de hormonas hipofisarias secundaria a 1 (deficiencia de ADH y GH).
Discusión
Los germinomas son neoplasias cerebrales primarias, diagnosticándose el 68% entre los 10 y 21 años
Los tumores de células germinales (TCG) pueden presentarse como neoplasias cerebrales primarias, principalmente en la edad pediátrica. La incidencia varía de un 0,3-0,5% de las neoplasias intracraneales primarias y aproximadamente un 3% de los tumores cerebrales primarios. El 68% de los germinomas se diagnostican entre los 10 y los 21 años, con un pico a los 10-12 años. Progresan rápidamente y se diseminan por el espacio subaracnoideo mediante infiltración periventricular. Asientan más frecuentemente en la glándula pineal (50-60%) y afectan más a varones (2,5/1), seguida del hipotálamo (30-40%), y otras localizaciones (ganglios de la base y tálamo). Cuando la afectación es pineal y supraselar se denominan germinomas bifocales.
El síntoma inicial suele ser una diabetes insípida durante meses
El síntoma inicial suele ser una diabetes insípida, en la cual la poliuria y la polidipsia pueden ser los únicos síntomas durante meses. Aparece más frecuentemente en aquellos tumores que afectan al infundíbulo (craneofaringioma, germinoma y glioma óptico), así como tras la cirugía (Tabla 1).
Las manifestaciones clínicas dependen de la localización. Los tumores pineales tienden a manifestarse con hidrocefalia y alteraciones del movimiento ocular (síndrome de Parinaud), y las lesiones de la región supraselar producen alteraciones visuales y alteraciones endocrinológicas, tales como diabetes insípida, retraso de crecimiento y pubertad precoz o retrasada, entre otras.
El déficit de GH es la complicación más frecuente, precoz y mejor estudiada de la radioterapia craneal, apareciendo entre el 52-100% de los pacientes. El déficit total de GH aparece con dosis >30Gy (aparece antes cuanto mayor dosis de radiación). Esta disfunción es a nivel hipotalámico, por alteración de los mecanismos de contrarregulación gobernados por el sistema GHRH/somatostatina dado que es más radiosensible que la hipófisis; dañándose ésta última sólo con dosis más altas de radiación.
El déficit de hormona de crecimiento es la complicación más frecuente y precoz tras la radioterapia
La talla final en nuestro paciente esta dentro de la normalidad para su talla familiar. La talla final inferior a la talla diana observada en algunos pacientes podría deberse a: déficit de GH, daño de la radioterapia sobre columna y huesos largos, malnutrición por disminución de la ingesta, recidiva tumoral, quimioterapia, corticoterapia, pubertad precoz y otras endocrinopatías. Existe asociación entre talla baja adulta y una menor edad al diagnóstico.
El tratamiento consiste en la administración de quimioterapia y radioterapia
Los tumores germinales son altamente radiosensibles y la radioterapia sola ha sido el tratamiento tradicional. En el pasado se utilizó la irradiación craneoespinal con refuerzo en la región del tumor primario, observándose una supervivencia a 5 años de un 90%. Actualmente se tiende a disminuir la dosis para minimizar los efectos secundarios. La quimioterapia (ciclofosfamida, etopósido, cisplatino y carboplatino) como tratamiento aislado tiene una tasa de respuesta total cercana al 90%, pero tiene un índice de recaídas del 50% y una toxicidad elevada, por lo que se utiliza como coadyuvante a la RT, lo que parece reducir su dosis y el volumen radiado con resultados óptimos.
Tablas y figuras
Tabla 1. Síntomas dependiendo de la localización
|
Localización |
Síntomas |
|
Región PINEAL |
Hidrocefalia Síndrome de Parinaud Signos Piramidales Ataxia |
|
Región SUPRASELAR |
Diabetes insípida Defectos visuales Hipopituitarismo Retraso puberal Deficiencia aislada de GH |
Figura 1.
Bibliografía
1. Aslan IR, Cheung CC. Early and late endocrine effects in pediatric central nervous system diseases. J Pediatr Rehabil Med. 2014;7:281-94
2. Wang Y, Zou L, Gao B. Intracranial germinoma: clinical and MRI findings in 56 patients. Childs Nerv Syst. 2010;26:1773-7.
3. Yang P, Li L, Kuang W, Li B, Zhou B, Yang J, Huang H. Intracranial multiple germ cell tumors: a case report and review of literature. Int J Clin Exp Pathol. 2014;7:9002-7
4. Al-Mahfoudh R1, Zakaria R, Irvine E, Pizer B, Mallucci CL. The management of bifocal intracranial germinoma in children. Childs Nerv Syst. 2014;30:625-30.
5. Jorsal T, Rørth M. Intracranial germ cell tumours. A review with special reference to endocrine manifestations. Acta Oncol. 2012;51:3-9.
6. Paximadis P, Hallock A, Bhambhani K, Chu R, Sood S, Wang Z, Konski A. Patterns of failure in patients with primary intracranial germinoma treated with neoadjuvant chemotherapy and radiotherapy. Pediatr Neurol. 2012;47:162-6.
7. Hu M, Guan H, Lau CC, Terashima K, Jin Z, Cui L et al. An update on the clinical diagnostic value of β-hCG and αFP for intracranial germ cell tumors. Eur J Med Res. 2016; 12;21:10.
8. Jensen AW, Laack NN, Buckner JC, Schomberg PJ, Wetmore CJ, Brown PD. Long-term follow-up of dose-adapted and reduced-field radiotherapy with or without chemotherapy for central nervous system germinoma. Int J Radiat Oncol Biol Phys. 2010 1;77:1449-56.

